7 Properties of Entropy
Part II — The Second Law
Last updated: 14-06-2026
7.1 Entropy of the ideal gas
We start from the simplest case and compute the entropy of an ideal gas directly from its definition (Definition 6.1) and the First Law, before any general machinery is in place. The result serves as the running example for the rest of the lecture.
The entropy was defined in Lecture 6 by Equation 6.8: relative to a reference state, the entropy of a state is the integral of \(\delta Q_{\rm rev}/T\) along any reversible quasi-static path connecting the two, and its value does not depend on the path. The heat \(\delta Q_{\rm rev}\) is itself path dependent, \(Q\) being no state function; what is path independent, and so defines a state function, is the integral of \(\delta Q_{\rm rev}/T\). We exploit this freedom by choosing a convenient path.
Take the reference state to be the gas at \((T_0, V_0)\), write \(S_0\) for its entropy, and let \(S(T, V)\) be the entropy of a generic state \((T, V)\). Then Equation 6.8 reads
\[S(T, V) = S_0 + \int_{\gamma} \frac{\delta Q_{\rm rev}}{T},\]
for any reversible quasi-static path \(\gamma\) from \((T_0, V_0)\) to \((T, V)\). We take \(\gamma\) to be an isothermal leg chained to an isochoric one:
- isothermal expansion at \(T_0\), from \(V_0\) to \(V\); then
- isochoric heating at \(V\), from \(T_0\) to \(T\).
The path is shown in the \((V, p)\) plane in Figure 7.1: leg (i) runs along the \(T_0\) isotherm to the intermediate state \((T_0, V)\), leg (ii) rises at fixed volume to the final state \((T, V)\).
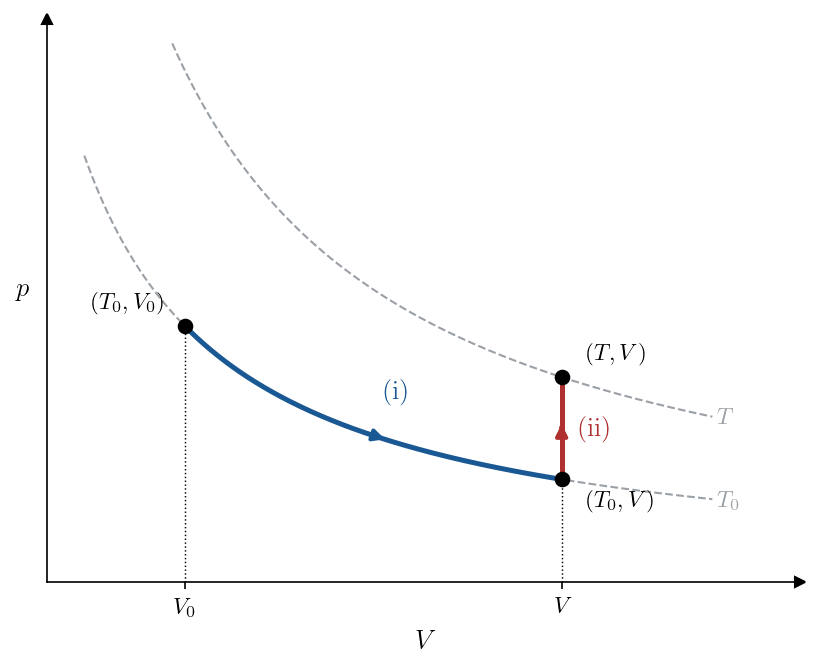
On either leg the reversible heat is fixed by the First Law, \(\delta Q_{\rm rev} = dU + p\,dV\) (Lecture 2), with \(dU = n\,c_V(T)\,dT\) for the ideal gas (Lecture 4), where the molar heat capacity \(c_V(T)\) may itself depend on temperature, and \(p = nRT/V\).
On the isothermal leg the temperature is held at \(T_0\), so \(dU = 0\) and \(\delta Q_{\rm rev} = p\,dV = (nRT_0/V)\,dV\); dividing by the temperature \(T_0\) at which the heat is exchanged,
\[\int_{(\mathrm i)} \frac{\delta Q_{\rm rev}}{T} = \int_{V_0}^{V} \frac{nRT_0/V'}{T_0}\,dV' = nR\ln\frac{V}{V_0}.\]
On the isochoric leg the volume is held fixed, so \(dV = 0\) and \(\delta Q_{\rm rev} = dU = n\,c_V(T)\,dT\); dividing by the running temperature \(T'\),
\[\int_{(\mathrm{ii})} \frac{\delta Q_{\rm rev}}{T} = n\int_{T_0}^{T} c_V(T')\,\frac{dT'}{T'}.\]
Adding the two legs gives the entropy of the ideal gas,
\[S(T,V) = S_0 + n\int_{T_0}^{T} c_V(T')\,\frac{dT'}{T'} + nR\ln\frac{V}{V_0}, \tag{7.1}\]
with \(S_0 = S(T_0, V_0)\). As with the internal energy, the derivation fixes only entropy differences: the constant \(S_0\) is left open, to be fixed by the Third Law later in the course. Chaining the two legs in the opposite order, an isochoric heating at \(V_0\) followed by an isothermal expansion at \(T\), produces the very same two integrals, so the sum is unchanged, exactly as the path independence built into Equation 6.8 requires (Problem 6.13).
For a gas whose molar heat capacity may be treated as constant over the temperature range of interest, the integral is elementary. This holds within any of the plateaus of Lecture 4: for instance a monatomic gas, with \(c_V \approx \tfrac{3}{2}R\) over a wide range, or a diatomic gas near room temperature, with \(c_V \approx \tfrac{5}{2}R\). The entropy then takes the closed form
\[S(T,V) = S_0 + n\,c_V\ln\frac{T}{T_0} + nR\ln\frac{V}{V_0}. \tag{7.2}\]
Entropy of an ideal gas — relative to a reference state \((T_0, V_0)\), \(S(T,V) = S_0 + n\!\int_{T_0}^{T} c_V(T')\,dT'/T' + nR\ln(V/V_0)\). For constant \(c_V\): \(S_0 + n\,c_V\ln(T/T_0) + nR\ln(V/V_0)\).
7.2 The increase of entropy
The entropy was defined in Lecture 6 through reversible processes alone: Equation 6.9 fixes the difference \(S(B)-S(A)\) from the heat exchanged along any reversible path between the two states. Most processes in nature are not reversible, and it is for these that the entropy has the most to say. We now bound the entropy difference for an arbitrary process.
The goal. Consider a system carried from an equilibrium state \(A\) to an equilibrium state \(B\) by some process that need not be reversible: it may be sudden, the system may fall out of equilibrium along the way, and it may possess no single temperature while it proceeds. Throughout, the system is in thermal contact with one reservoir at temperature \(T_{\rm res}\), from which it absorbs a total heat \(Q_{\rm irr}\). We want to bound \(S(B)-S(A)\) in terms of \(Q_{\rm irr}\) and \(T_{\rm res}\).
Closing the cycle. Return the system from \(B\) to \(A\) along a reversible quasi-static path, chosen reversible on purpose so that the heat it exchanges is controlled by an entropy difference we already know. The two legs together, the given process \(A\to B\) and the reversible return \(B\to A\), form a cyclic process (Figure 7.2), to which we apply the first Clausius inequality (Theorem 6.2). That inequality sums the heat the cycle absorbs from each reservoir in its surroundings, divided by that reservoir’s temperature, and bounds the total by zero. We tally the two legs in turn.
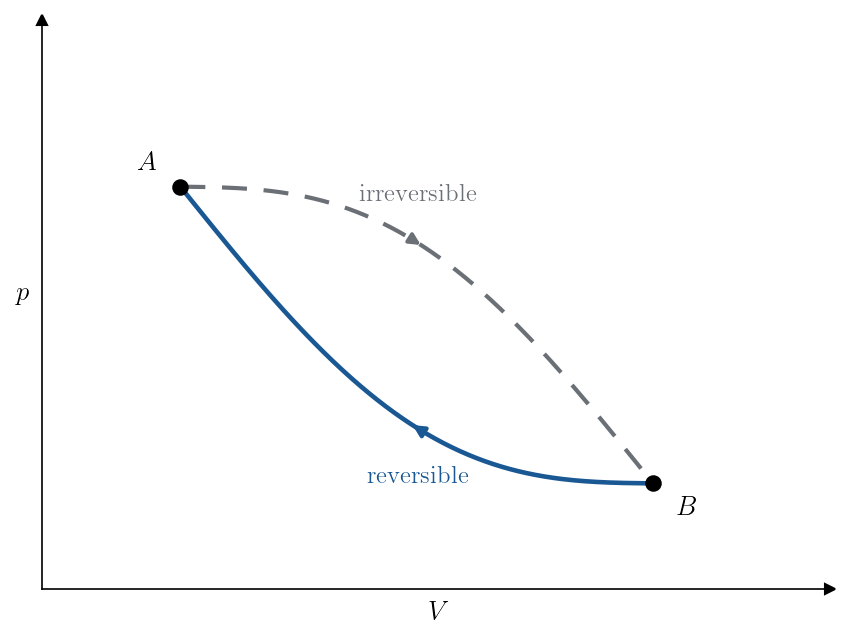
The irreversible leg. The system exchanges heat with the single reservoir at \(T_{\rm res}\), absorbing \(Q_{\rm irr}\) in all, so this leg contributes a single term \(Q_{\rm irr}/T_{\rm res}\) to the Clausius sum. Here \(T_{\rm res}\) is the reservoir’s temperature: the system’s own temperature need not be defined at any point of this leg, and does not enter. This is exactly the setting the first Clausius inequality was built for, since it refers only to the reservoirs.
The reversible leg. For this leg we repeat the construction behind the second Clausius inequality (Lecture 6, Theorem 6.3). Subdivide the reversible return into \(n\) steps. Because the leg is quasi-static, in the \(k\)-th step the system is in equilibrium at a well-defined temperature \(T_k\), and its surroundings differ from it only infinitesimally, so the step amounts to exchanging a small heat \(\delta Q_k\) with a reservoir at that same temperature \(T_k\). These steps contribute \(\sum_{k} \delta Q_k/T_k\) to the Clausius sum, and as the subdivision is refined the Riemann sum converges to the line integral \(\int_{B\to A}^{\rm rev}\delta Q_{\rm rev}/T\), in which \(T\) is now the system’s own temperature at each point of the path.
Assembling the cycle. The cycle therefore exchanges heat with the reservoir at \(T_{\rm res}\) on the first leg and with the sequence of reservoirs at temperatures \(T_k\) on the second. Applying the first Clausius inequality to this whole collection of reservoirs,
\[\frac{Q_{\rm irr}}{T_{\rm res}} + \int_{B\to A}^{\rm rev}\frac{\delta Q_{\rm rev}}{T} \le 0 .\]
Along the reversible return the line integral is fixed by the definition of entropy, \(\int_{B\to A}^{\rm rev}\delta Q_{\rm rev}/T = S(A)-S(B)\) (Equation 6.9). Substituting and rearranging gives the open-path Clausius inequality,
\[S(B) - S(A) \ge \frac{Q_{\rm irr}}{T_{\rm res}} , \tag{7.3}\]
valid for any process carrying the system from \(A\) to \(B\) while in contact with a reservoir at \(T_{\rm res}\), with equality if and only if that process is itself reversible. The temperature on the right is the reservoir’s, \(T_{\rm res}\), not the system’s. The distinction is real: on the irreversible leg the system may be out of equilibrium and have no single temperature at all, while the reservoir it draws heat from always has one. Only when the process is reversible do the two coincide at every step, the heat becomes \(\delta Q_{\rm rev}\) exchanged at the system’s own temperature, and the inequality closes to the equality that defines the entropy.
More than one reservoir. If the system exchanges heat with several reservoirs as it goes from \(A\) to \(B\), absorbing \(Q_j\) from a reservoir at temperature \(T_{{\rm res},j}\) for \(j = 1,\ldots,m\), the irreversible leg contributes \(\sum_j Q_j/T_{{\rm res},j}\) to the Clausius sum, and the same argument bounds the entropy difference by that sum,
\[S(B) - S(A) \ge \sum_{j=1}^{m} \frac{Q_j}{T_{{\rm res},j}} . \tag{7.4}\]
Each \(T_{{\rm res},j}\) is again a reservoir temperature, and equality holds only if every exchange is reversible. (When the surroundings’ temperature varies continuously, the sum passes to a line integral \(\int_{A\to B}\delta Q/T_{\rm res}\) over the actual process, with \(T_{\rm res}\) the temperature of whatever supplies the heat \(\delta Q\) at each stage.)
The isolated system. The statement is sharpest for an isolated system, which exchanges no heat with anything: every \(Q_j = 0\), and Equation 7.4 reduces to
\[\Delta S_{\rm isolated} = S(B) - S(A) \ge 0 . \tag{7.5}\]
This is the law of increase of entropy: the entropy of an isolated system never decreases, and holds steady only if every process within it is reversible. Since any system together with its surroundings forms an isolated whole, \(\Delta S_{\rm univ} = \Delta S_{\rm sys} + \Delta S_{\rm surr} \ge 0\), so a decrease in one part must be at least matched by an increase in another. This is the entropy form of the Second Law, and, unlike the mechanical laws beneath it, the inequality is not symmetric under reversal of time: it is what gives a thermodynamic process a direction.
The free expansion of an ideal gas makes the law concrete. Let \(n\) moles expand from \(V_1\) to \(V_2\) into an evacuated, rigid, insulated vessel. The gas pushes on nothing, so no work is done; the vessel is insulated, so no heat enters, \(Q = 0\); hence \(U\), and with it \(T\), is unchanged, and Equation 7.1 gives
\[\Delta S = nR\ln\frac{V_2}{V_1} > 0 .\]
The gas is isolated, yet its entropy has risen, exactly as Equation 7.5 requires. Because the vessel is insulated no heat crosses its walls and no reservoir temperature enters Equation 7.5 at all; the \(T\) in \(\Delta S\) above is the gas’s own, appearing only through the state function Equation 7.1. No heat was driven in and no work taken out; the increase was produced entirely within, by the irreversibility of the expansion. Heat flow across a finite temperature difference (the problems of this lecture) and the mixing of gases (Lecture 11) are further such processes; each raises \(\Delta S_{\rm univ}\).
Finally, the law selects the equilibrium state of an isolated system. Released from a constraint, the system evolves until no further increase of entropy is possible, so equilibrium is the state of maximum entropy compatible with the constraints. Turning this into explicit conditions for thermal, mechanical, and chemical equilibrium between subsystems is the subject of Lecture 8.
7.3 Entropy in differential form
The relations so far have been stated for finite changes, between two equilibrium states \(A\) and \(B\). For an infinitesimal process, carrying the system between two neighbouring equilibrium states, they take a differential form, more compact and better suited to combining with the First Law in the next section.
Because the entropy is a state function, it possesses a well-defined infinitesimal change \(dS\) between an equilibrium state and a neighbouring one. Unlike \(\delta Q\), the heat absorbed in a particular infinitesimal process, which is not the increment of any state function, \(dS\) is an exact differential: its integral between two equilibrium states depends only on the endpoints,
\[\int_A^B dS = S(B) - S(A),\]
whatever the path joining them. We now write the relations of the previous sections for a single infinitesimal process, taking care, at each one, of whether the temperature that appears is the system’s or a reservoir’s.
The definition of entropy. In a reversible infinitesimal process the system stays in equilibrium at its own temperature \(T\) and absorbs a heat \(\delta Q_{\rm rev}\) at that temperature. The definition of entropy (Equation 6.9) then reads, in differential form,
\[dS = \frac{\delta Q_{\rm rev}}{T} \qquad (\text{reversible infinitesimal process}), \tag{7.6}\]
where \(T\) is the temperature of the system. Equivalently \(\delta Q_{\rm rev} = T\, dS\). The heat \(\delta Q_{\rm rev}\) is inexact, yet dividing it by the system temperature \(T\) yields the exact differential \(dS\); we return to this, as the integrating factor \(1/T\), when reading the fundamental relation as a relation among state functions. For the ideal gas, Equation 7.1 is the integral of
\[dS = n\,c_V(T)\,\frac{dT}{T} + nR\,\frac{dV}{V}.\]
The Clausius inequality. In an arbitrary infinitesimal process, in which the system absorbs a heat \(\delta Q\) from a reservoir at temperature \(T_{\rm res}\), the open-path inequality Equation 7.3 becomes
\[dS \ge \frac{\delta Q}{T_{\rm res}}, \tag{7.7}\]
with equality if and only if the infinitesimal process is reversible. The contrast with Equation 7.6 lies precisely in the temperature: here \(T_{\rm res}\) is the temperature of the reservoir that supplies the heat, not that of the system, which in an irreversible infinitesimal process may have no single value at all. Only when the process is reversible do the two temperatures coincide, \(\delta Q\) becomes \(\delta Q_{\rm rev}\), and Equation 7.7 closes to the equality Equation 7.6.
The isolated system. For an isolated system \(\delta Q = 0\), and Equation 7.7 gives
\[dS \ge 0,\]
the differential form of the law of increase of entropy (Equation 7.5): the entropy of an isolated system cannot decrease in any infinitesimal process.
7.4 Work and the boundary pressure
Work is energy in transit across the system boundary, so what governs it is the force acting at that boundary. In an infinitesimal process the work a system delivers as it moves its boundary is
\[\delta W = p_{\rm ext}\,dV,\]
where \(p_{\rm ext}\) is the pressure at the boundary, against which the system pushes as it sweeps out a volume \(dV\) (the external pressure of Lecture 2). It is the boundary pressure that enters, because the energy leaving the system is reckoned at the boundary: by Newton’s third law the system and its surroundings push on each other with equal and opposite forces across the contact surface, and the energy delivered is that force times the displacement of the surface, \(p_{\rm ext}\,dV\). The surroundings always exert a definite pressure there, even during a violent process in which the system is non-uniform and has no single pressure of its own. So \(p_{\rm ext}\), and not any internal value, is the quantity that always makes sense in \(\delta W\).
This refines, rather than contradicts, the \(\delta W = p\,dV\) of Lecture 2. There \(p\) was introduced deliberately as the pressure the gas exerts at the moving boundary, which equals the bulk thermodynamic pressure only while the gas stays uniform; that same discussion noted that for a rapid process the work is governed instead by the external pressure, and deferred the general case to here. The boundary pressure \(p_{\rm ext}\) is precisely that quantity, now named and studied in its own right. Whether it equals the gas pressure \(p\) is a separate question, and two things bear on it: the gas has a single, well-defined pressure \(p\) only when it is in internal equilibrium; and the boundary, hence \(p_{\rm ext}\), depends on what we choose as the system (Figure 7.3). The examples below show when \(p_{\rm ext}\) equals the gas pressure \(p\) and when it does not.
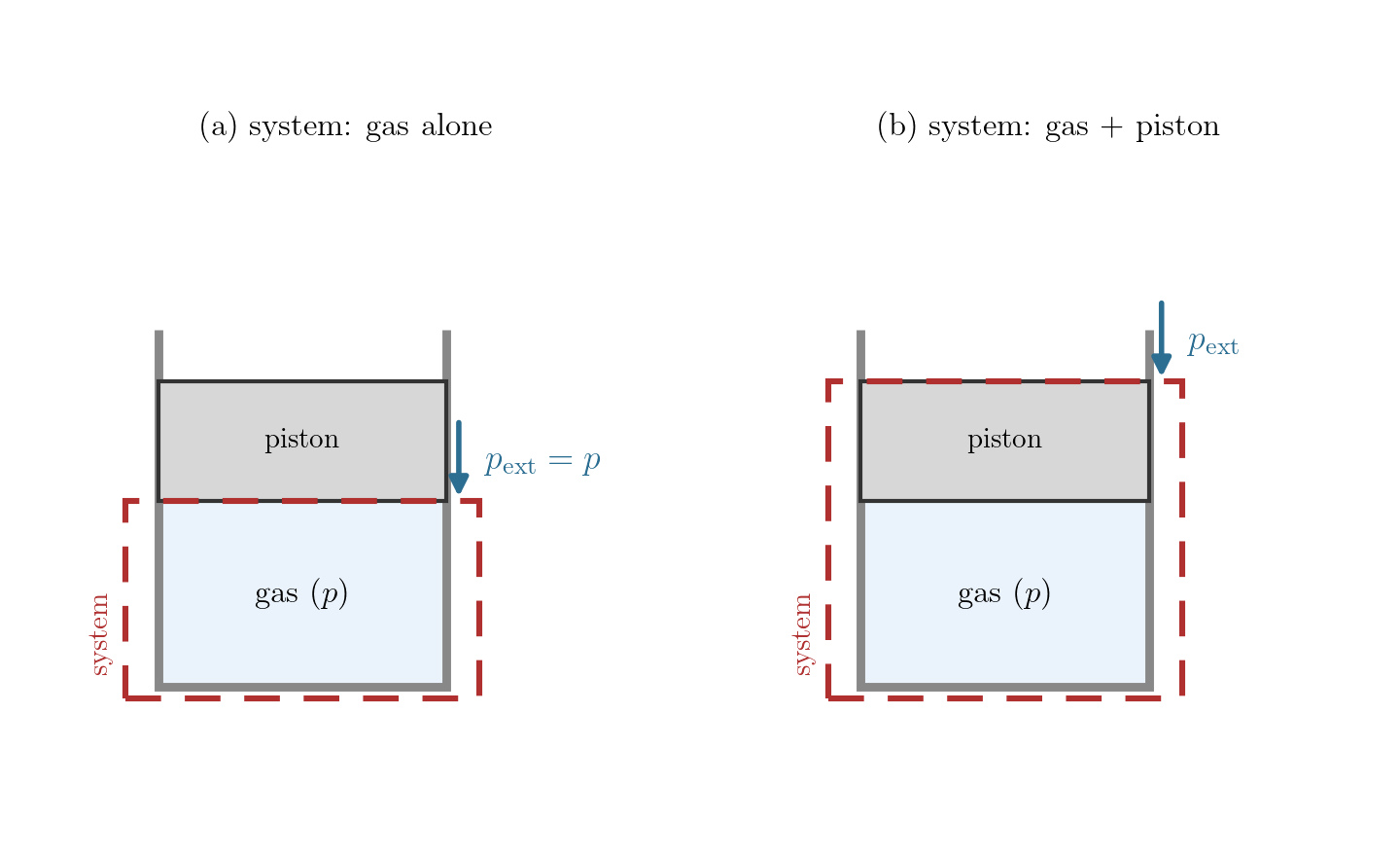
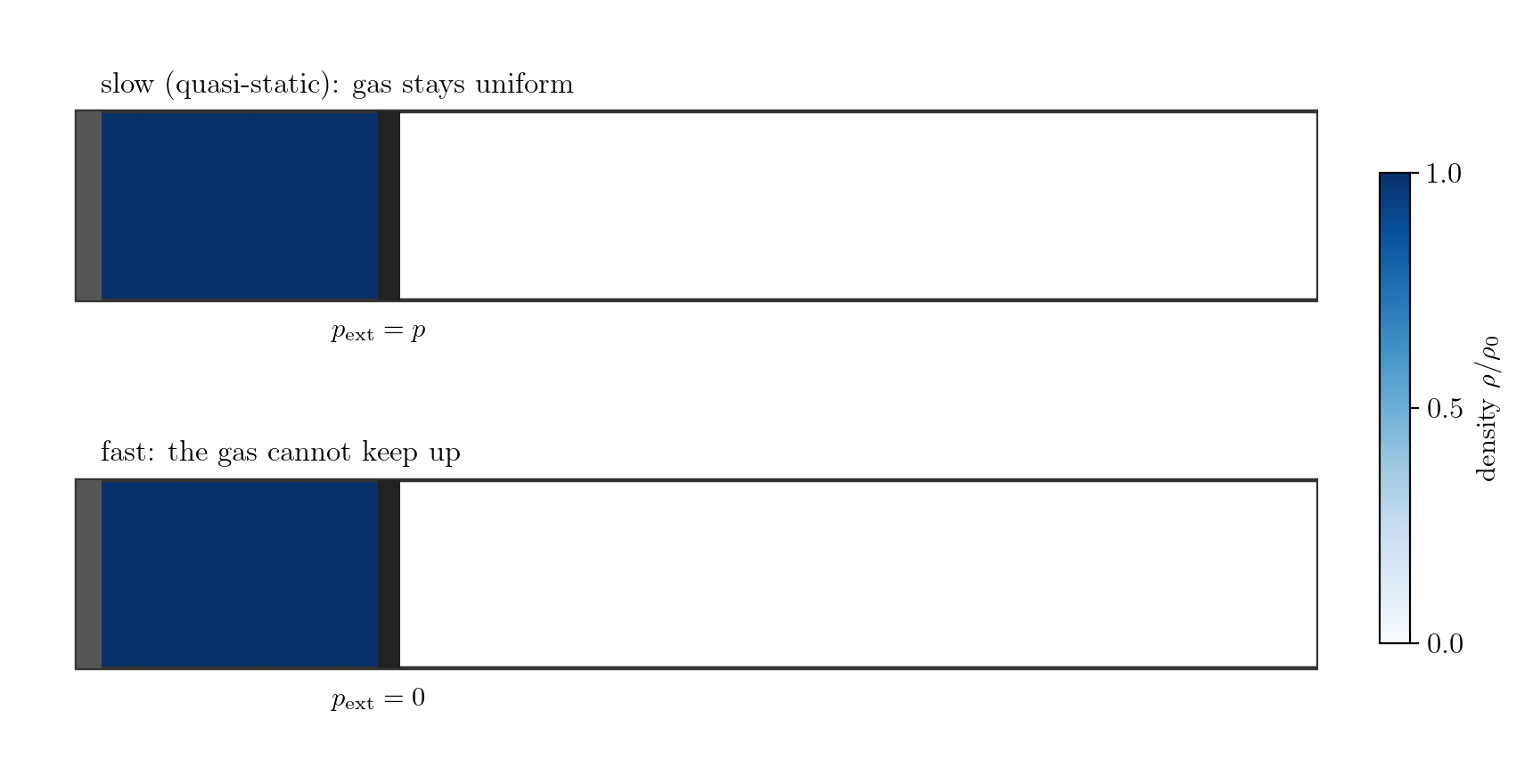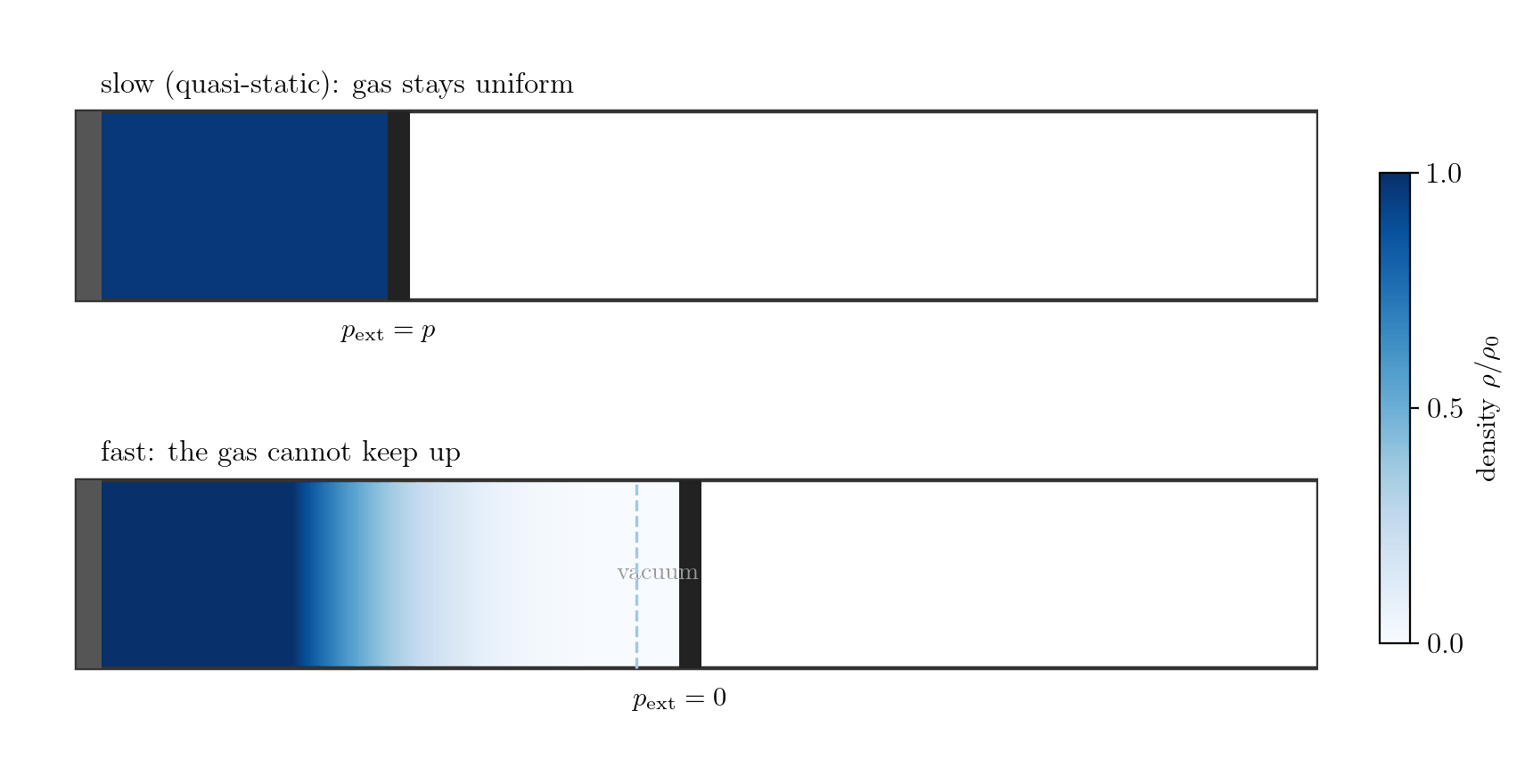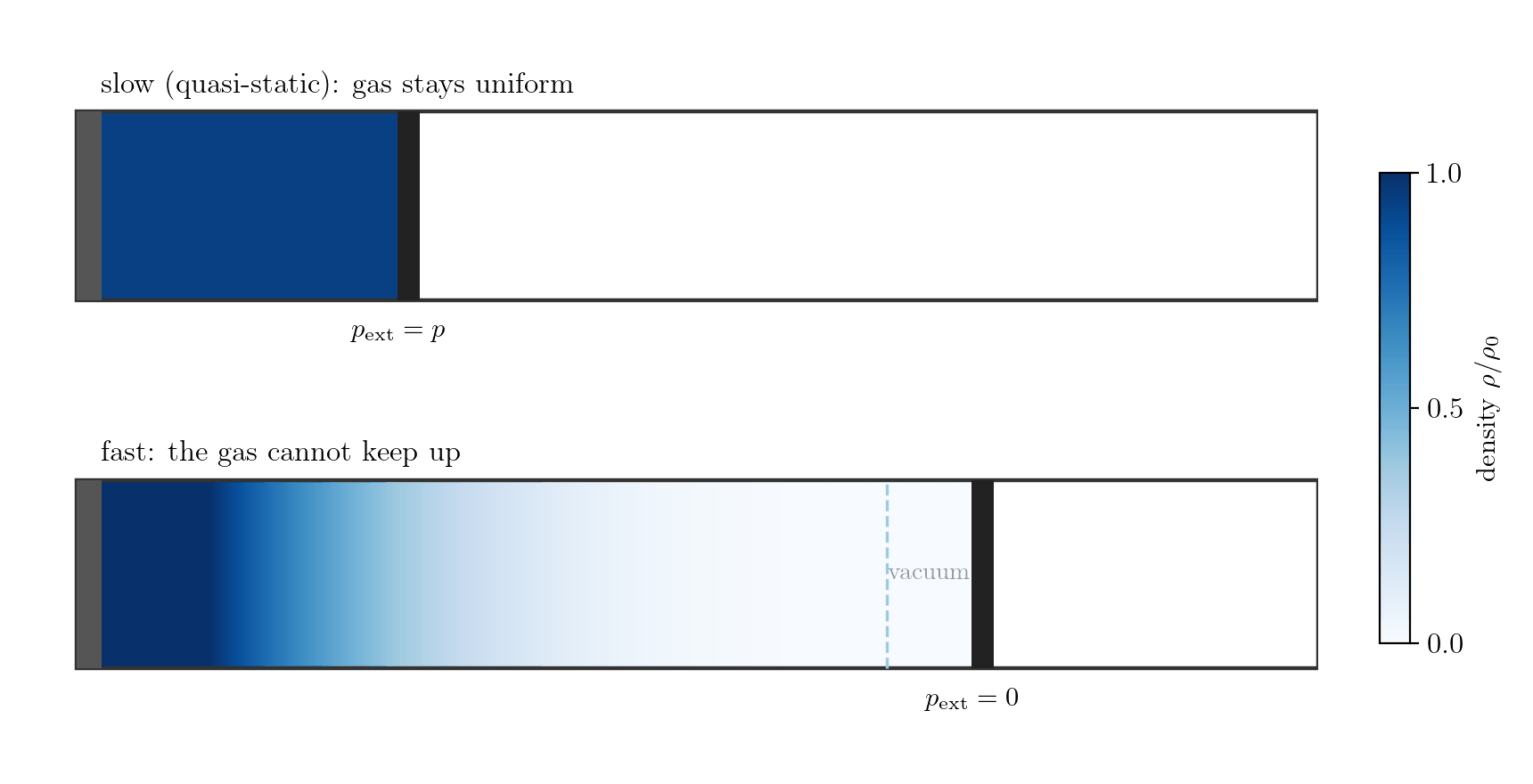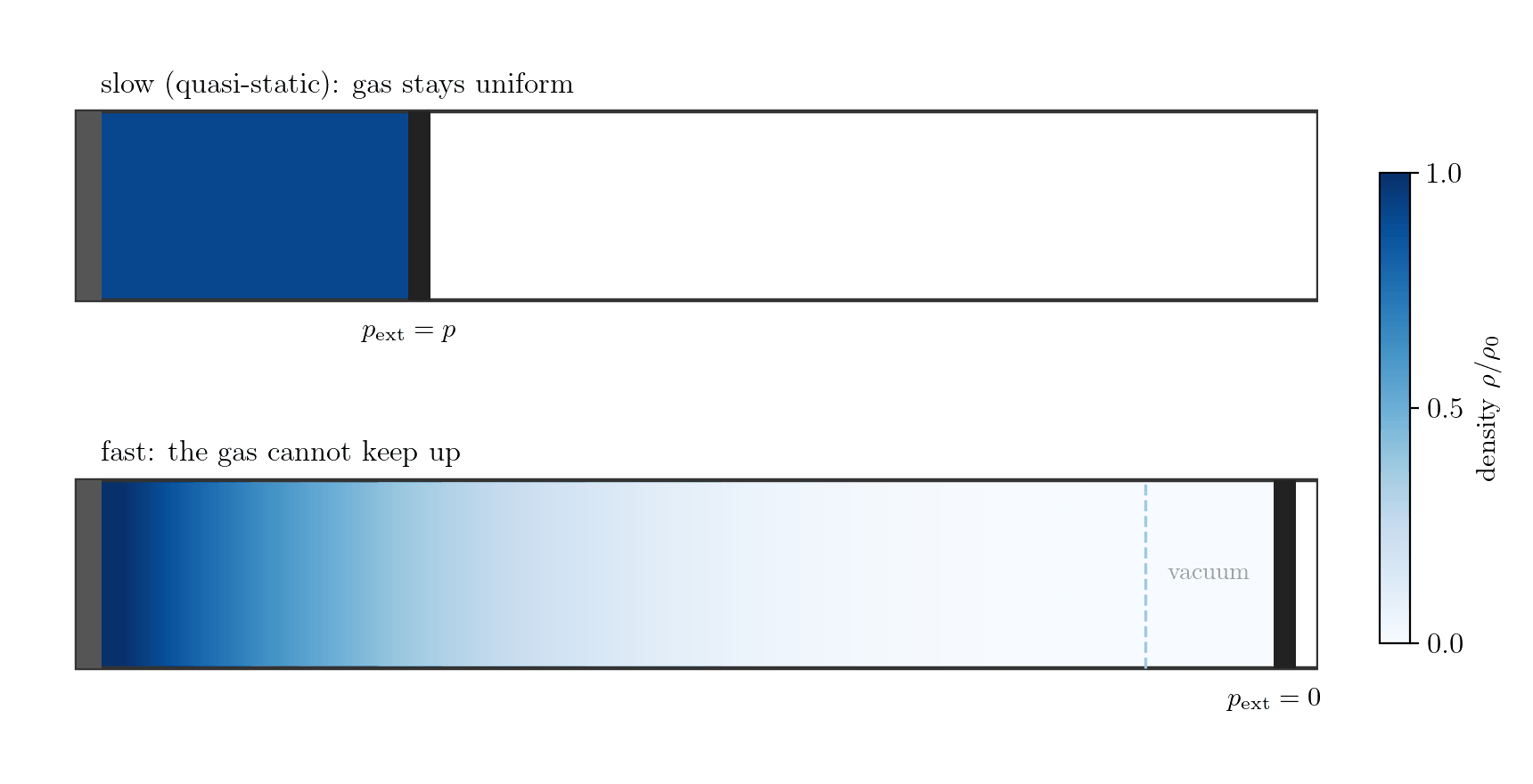
Example 1: the gas alone, expanding fast. Let the system be the gas in a cylinder closed by a piston; the boundary is the gas-piston interface and \(p_{\rm ext}\) is the pressure the gas exerts on the piston. Moved slowly, the gas stays uniform, the interface pressure is the bulk pressure, \(p_{\rm ext} = p\), and \(\delta W = p\,dV\). Withdraw the piston faster than the gas can follow, however, and the gas rarefies and loses contact with it (Figure 7.4): a vacuum forms at the face, so the gas does no work there, \(p_{\rm ext}=0\) and \(\delta W = 0\), while the bulk gas is non-uniform and has no single pressure \(p\) at all. Losing internal equilibrium destroys both the equality \(p_{\rm ext}=p\) and the meaning of \(p\) itself; this is free expansion.
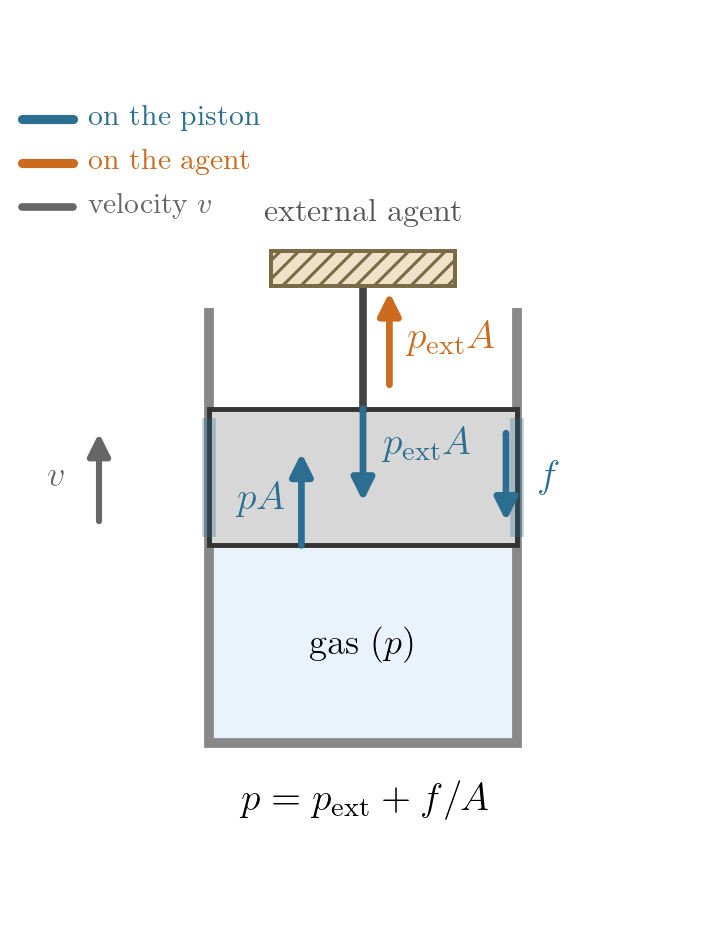
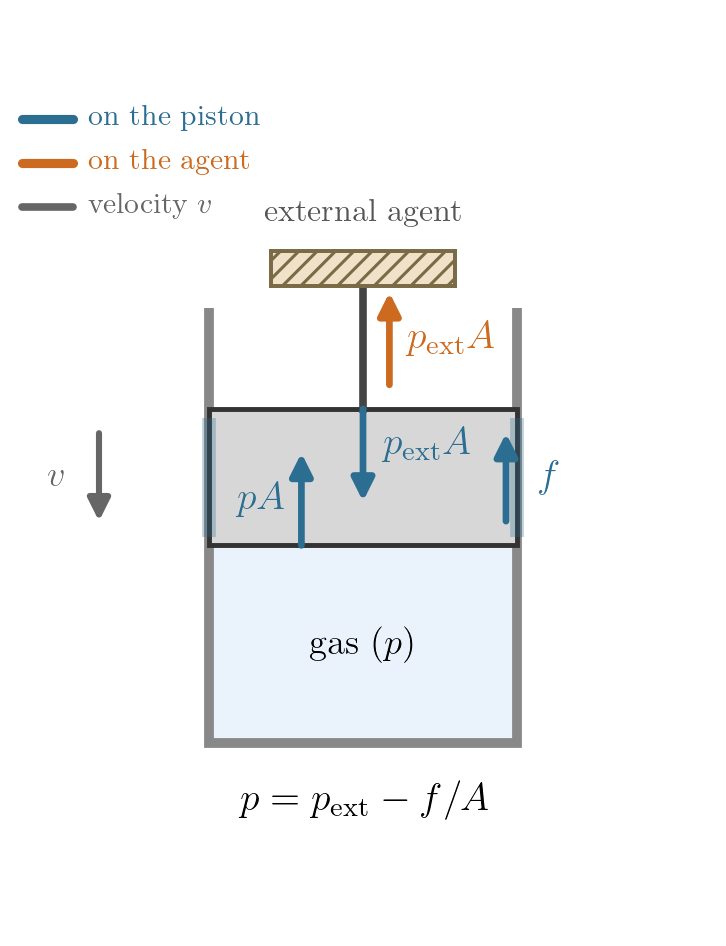
Example 2: gas and piston, with friction. Now include the piston in the system, so the boundary is its outer face and \(p_{\rm ext}\) is the pressure applied there from outside. Let the piston (of area \(A\)) slide against the cylinder with a constant friction force of magnitude \(f\), and take the process quasi-static, so the gas is uniform and \(p\) is well defined. Friction always opposes the piston’s motion, so its direction, and with it the force balance, depends on which way the piston travels (Figure 7.5). On expansion the piston moves out and friction acts inward,
\[pA = p_{\rm ext}A + f \quad\Longrightarrow\quad p = p_{\rm ext} + \frac{f}{A};\]
on compression the piston moves in, friction acts outward,
\[pA + f = p_{\rm ext}A \quad\Longrightarrow\quad p = p_{\rm ext} - \frac{f}{A}.\]
In both cases \(p \neq p_{\rm ext}\), but the sign of the gap reverses with the direction of travel. The work delivered to the outside is \(\delta W = p_{\rm ext}\,dV\), while the gas does \(p\,dV\) on the piston; the difference \((p - p_{\rm ext})\,dV = f\,|dx| \ge 0\) is dissipated by friction whichever way the piston moves, since \(p - p_{\rm ext}\) and \(dV\) change sign together (on compression both turn negative, so their product stays positive). So \(p\) is perfectly well defined, yet it is not the pressure that does the external work, and the process, though quasi-static, is irreversible in either direction: the gas stays in equilibrium throughout, but friction dissipates whichever way the piston moves, so being quasi-static does not make it reversible.
Example 3: gas and piston, frictionless. Remove the friction. The same balance gives \(pA = p_{\rm ext}A\), hence \(p = p_{\rm ext}\) and \(\delta W = p\,dV = p_{\rm ext}\,dV\), with nothing dissipated. This is the reversible case, in which gas pressure and boundary pressure coincide.
What the examples establish. The work is set by the boundary pressure \(p_{\rm ext}\), and whether this equals the gas pressure \(p\) depends on where the system boundary is drawn. For the gas alone, \(p_{\rm ext}=p\) whenever the gas is in internal equilibrium, and Example 1 is the only way it fails. Enlarging the system to include the piston moves the boundary to its outer face; there \(p_{\rm ext}=p\) only for an ideal, massless and frictionless piston (Example 3), whereas friction makes \(p_{\rm ext}\neq p\) and dissipates the difference (Example 2). The fundamental relation concerns the gas, carried through equilibrium states, for which \(\delta W = p\,dV\); this is what we combine with the Second Law next.
7.5 Heat, work, and entropy in an infinitesimal process
We now bring the First and Second Laws together for a single infinitesimal process of a closed system. The result, read off in the next section, is a relation among state functions; here we keep the process in view and bound heat, work, and entropy separately.
For any infinitesimal process the First Law reads
\[dU = \delta Q - \delta W = \delta Q - p_{\rm ext}\, dV,\]
with \(\delta W = p_{\rm ext}\, dV\) the boundary work of the preceding section. The process need not be reversible, nor the system even in equilibrium: \(\delta Q\) is the heat absorbed from a reservoir at temperature \(T_{\rm res}\), \(p_{\rm ext}\) is the boundary pressure, and the system has a temperature \(T\) and a pressure \(p\) of its own only while it is in internal equilibrium. The Second Law enters through the Clausius inequality Equation 7.7, \(dS \ge \delta Q/T_{\rm res}\), in which \(T_{\rm res}\) is again the reservoir’s temperature, not the system’s. The gap between the two sides defines the entropy generated by the process,
\[\delta S_{\rm gen} = dS - \frac{\delta Q}{T_{\rm res}} \ge 0, \tag{7.8}\]
the total entropy change of the system and the reservoir together (the reservoir’s entropy changes by \(-\delta Q/T_{\rm res}\)). It is non-negative always and zero exactly when the process is reversible: it measures the irreversibility of the process.
The work needs the same care over which pressure appears. The energy crossing the boundary is \(\delta W = p_{\rm ext}\, dV\), set by the boundary pressure. When the system is in internal equilibrium, so that its own pressure \(p\) is defined, the boundary can advance only if the gas pushes at least as hard as the resistance it meets: on expansion \(p \ge p_{\rm ext}\) with \(dV > 0\), on compression \(p \le p_{\rm ext}\) with \(dV < 0\), so in either direction
\[\delta W = p_{\rm ext}\, dV \le p\, dV, \tag{7.9}\]
the shortfall \((p - p_{\rm ext})\, dV \ge 0\) being the work lost to mechanical dissipation (the friction of the preceding section). The gas pressure \(p\) sets an upper bound on the work delivered, reached only at a mechanically balanced boundary, \(p_{\rm ext} = p\).
A reversible infinitesimal process is one for which \(\delta S_{\rm gen} = 0\), and this forces both equalities at once. It demands thermal equilibrium with the reservoir, \(T = T_{\rm res}\), so that the heat is \(\delta Q = T\, dS\) at the system’s own temperature (Equation 7.6); and mechanical equilibrium at the boundary, \(p_{\rm ext} = p\), so that \(\delta W = p\, dV\), the equality in Equation 7.9. Substituting both into the First Law gives
\[dU = T\, dS - p\, dV, \tag{7.10}\]
the fundamental relation of thermodynamics, here obtained along a reversible infinitesimal process.
It holds, in fact, for every process between the two equilibrium endpoints, because the heat and the work depart from \(T\, dS\) and \(p\, dV\) by the same amount, which cancels in \(dU\). While the system stays in internal equilibrium, \(T\, dS = dU + p\, dV\) between its states; eliminating \(dU\) with the First Law gives \(T\, dS = \delta Q + (p - p_{\rm ext})\, dV\), so
\[ \begin{aligned} \delta Q &= T\, dS - (p - p_{\rm ext})\, dV, \\ \delta W &= p\, dV - (p - p_{\rm ext})\, dV , \end{aligned} \]
the common shortfall \((p - p_{\rm ext})\, dV\) drops out of \(dU = \delta Q - \delta W\), leaving \(dU = T\, dS - p\, dV\) whatever the path. The same elimination splits the generated entropy Equation 7.8 into its two sources,
\[\delta S_{\rm gen} = \delta Q\left(\frac{1}{T} - \frac{1}{T_{\rm res}}\right) + \frac{(p - p_{\rm ext})\, dV}{T}, \tag{7.11}\]
valid while the system is in internal equilibrium, so that a single temperature \(T\) is defined: a thermal part, from heat crossing the finite gap between the system temperature \(T\) and the reservoir temperature \(T_{\rm res}\) (non-negative because, by the Second Law, heat flows from hot to cold), and a mechanical part, the dissipated work of Equation 7.9 divided by \(T\). Each vanishes only in its own equilibrium, \(T = T_{\rm res}\) and \(p = p_{\rm ext}\), together restoring reversibility. Over a finite process the generated entropy accumulates to \(\Delta S_{\rm gen} = \int \delta S_{\rm gen} \ge 0\), a measure of the departure from reversibility and, as the next example shows, of the work thereby lost.
Worked example: same endpoints, different heat and work. Take \(n\) moles of ideal gas at temperature \(T\), in contact with a reservoir at the same temperature \(T\), and expand it isothermally from \(V_1\) to \(V_2 = 2V_1\), so the gas pressure falls from \(p_1 = nRT/V_1\) to \(p_2 = nRT/V_2 = \tfrac{1}{2}p_1\). We drive it to this final state in three different ways. Since the end states are fixed, so are the state-function changes:
\[ \begin{aligned} \Delta U &= 0 \quad (\text{ideal gas, } \Delta T = 0), \\ \Delta S &= nR\ln\frac{V_2}{V_1} = nR\ln 2, \end{aligned} \]
so \(T\,\Delta S = nRT\ln 2\) in every case. What changes is the boundary pressure \(p_{\rm ext}\) against which the piston moves, and with it the work \(W = \int_{V_1}^{V_2} p_{\rm ext}\,dV\) and the heat \(Q = \Delta U + W = W\).
Slow expansion. The piston is let out slowly, the gas stays in equilibrium, and it presses on the piston with its own pressure, \(p_{\rm ext} = p = nRT/V\). Then
\[ \begin{aligned} W &= \int_{V_1}^{V_2}\frac{nRT}{V}\,dV = nRT\ln\frac{V_2}{V_1} = nRT\ln 2, \\ Q &= nRT\ln 2 = T\,\Delta S . \end{aligned} \]
The heat absorbed equals \(T\,\Delta S\); nothing is wasted (this is the reversible case).
Sudden expansion against a fixed pressure. The piston is released against a constant boundary pressure equal to the final gas pressure, \(p_{\rm ext} = p_2 = nRT/V_2 = \tfrac{1}{2}p_1\). While the gas pressure exceeds \(p_{\rm ext}\) the piston accelerates outward; once it falls below \(p_{\rm ext}\) the piston decelerates, and the gas comes to rest where the two pressures balance. Setting \(p_{\rm ext} = p_2\), the pressure of the target state, is what brings the gas to rest exactly at \(V_2\); against a higher pressure it would stop short. With \(p_{\rm ext}\) constant,
\[ \begin{aligned} W &= p_{\rm ext}\,(V_2 - V_1) = \frac{nRT}{V_2}(V_2 - V_1) = nRT\left(1 - \frac{V_1}{V_2}\right) = \frac{nRT}{2}, \\ Q &= \frac{nRT}{2} . \end{aligned} \]
Less work crosses the boundary, and less heat is drawn from the reservoir.
Free expansion. The piston is removed (the gas expands into an evacuated space), so it pushes on nothing, \(p_{\rm ext} = 0\), and
\[W = Q = 0.\]
Collecting the three cases (with \(n = 1\ \mathrm{mol}\) and \(T = 300\ \mathrm{K}\), so \(T\,\Delta S = nRT\ln 2 = 1729\ \mathrm{J}\)):
| Expansion | \(p_{\rm ext}\) | \(W\) | \(Q\) | lost work \(\;T\,\Delta S_{\rm gen} = T\,\Delta S - Q\) |
|---|---|---|---|---|
| slow (reversible) | \(p = nRT/V\) | \(1729\ \mathrm{J}\) | \(1729\ \mathrm{J}\) | \(0\) |
| against \(p_{\rm ext} = \tfrac{1}{2}p_1\) | \(\tfrac{1}{2}p_1\) | \(1247\ \mathrm{J}\) | \(1247\ \mathrm{J}\) | \(482\ \mathrm{J}\) |
| free | \(0\) | \(0\) | \(0\) | \(1729\ \mathrm{J}\) |
The change of state (\(\Delta U\), \(\Delta S\)) is the same in every row, while \(Q\) and \(W\) shrink together as \(p_{\rm ext}\) is lowered, exactly as \(Q = \Delta U + \int p_{\rm ext}\,dV\) requires. The shortfall \(T\,\Delta S - Q\) is the lost work \(T\,\Delta S_{\rm gen}\): zero for the slow expansion, and largest for the free one, which wastes the entire \(nRT\ln 2\).
7.6 The fundamental relation
The fundamental relation Equation 7.10,
\[dU = T\, dS - p\, dV,\]
was reached along a reversible process, but it contains only state functions. It is therefore a relation among the state functions of the system, holding between any two neighbouring equilibrium states irrespective of how, or whether, the system is actually taken between them. Read this way it is a statement about states, not about a process, and we now draw out its consequences.
The relation invites two complementary readings. In the first, the internal energy is regarded as a function of the entropy and the volume, \(U = U(S, V)\). Its total differential is \(dU = (\partial U/\partial S)_V\, dS + (\partial U/\partial V)_S\, dV\). Two expressions linear in the independent increments \(dS\) and \(dV\) can be equal for all \(dS\) and \(dV\) only if their coefficients agree one by one, so comparing this with Equation 7.10 identifies the two coefficients,
\[ \begin{aligned} T &= \left(\frac{\partial U}{\partial S}\right)_V, \\ p &= -\left(\frac{\partial U}{\partial V}\right)_S . \end{aligned} \tag{7.12}\]
So \(S\) and \(V\) are the natural variables of \(U\): the single function \(U(S, V)\) yields the temperature and pressure by differentiation.
In the second reading, the same relation is solved for the entropy,
\[dS = \frac{1}{T}\, dU + \frac{p}{T}\, dV, \tag{7.13}\]
and the entropy is regarded as a function of the energy and the volume, \(S = S(U, V)\). Matching with its total differential \(dS = (\partial S/\partial U)_V\, dU + (\partial S/\partial V)_U\, dV\) gives
\[ \begin{aligned} \left(\frac{\partial S}{\partial U}\right)_V &= \frac{1}{T}, \\ \left(\frac{\partial S}{\partial V}\right)_U &= \frac{p}{T} . \end{aligned} \tag{7.14}\]
The two readings carry the same content, related by inverting \(U(S, V)\) into \(S(U, V)\); their derivatives agree, since at fixed \(V\) the inverse-function rule gives \((\partial S/\partial U)_V = 1/(\partial U/\partial S)_V = 1/T\). This reciprocal holds only because the same variable \(V\) is held fixed on both sides, reducing it to a one-variable inverse; it would fail if different variables were held fixed. The identity \((\partial S/\partial U)_V = 1/T\) is what will fix thermal equilibrium between subsystems in Lecture 8. The factor \(1/T\) appearing here is an integrating factor: it turns the inexact reversible heat \(\delta Q_{\rm rev} = dU + p\, dV\) into the exact differential \(dS\). The ideal-gas entropy Equation 7.1 was an instance of this: although \(\delta Q_{\rm rev}\) is path dependent, the integral of \(\delta Q_{\rm rev}/T\) depends only on the endpoints, which is what allowed it to define the state function \(S(T, V)\).
The \(T\, dS\) equation. A practical form follows on choosing the temperature and the volume as independent variables. Writing \(U = U(T, V)\), its differential is \(dU = (\partial U/\partial T)_V\, dT + (\partial U/\partial V)_T\, dV\); substituting into Equation 7.10,
\[T\, dS = n\,c_V\, dT + \left[\left(\frac{\partial U}{\partial V}\right)_T + p\right] dV, \tag{7.15}\]
where \(n\,c_V = (\partial U/\partial T)_V\) is the heat capacity at constant volume (Lecture 3), in general a function of temperature. For an ideal gas \((\partial U/\partial V)_T = 0\) and \(p = nRT/V\), giving \(T\, dS = n\,c_V\, dT + (nRT/V)\, dV\), which on dividing by \(T\) reproduces Equation 7.1 in one step. The coefficient \((\partial U/\partial V)_T\) measures the departure from ideal-gas behaviour and is evaluated for real substances in Lecture 9.
7.7 Summary
For the ideal gas the entropy follows from the First Law and the definition of entropy: relative to a reference state \((T_0, V_0)\), \(S(T,V)= S_0 + n\!\int_{T_0}^{T} c_V(T')\,dT'/T' + nR\ln(V/V_0)\) (Equation 7.1), reducing to \(S_0 + n\,c_V\ln(T/T_0)+nR\ln(V/V_0)\) for constant \(c_V\) (Equation 7.2).
The open-path Clausius inequality \(S(B)-S(A)\ge Q_{\rm irr}/T_{\rm res}\) (Equation 7.3), with \(T_{\rm res}\) the temperature of the reservoir supplying the heat, generalizes to several reservoirs as \(S(B)-S(A)\ge\sum_j Q_j/T_{{\rm res},j}\) (Equation 7.4) and gives the law of increase of entropy: for an isolated system \(\Delta S\ge 0\) (Equation 7.5), with equality only for reversible change.
For an infinitesimal process the entropy has an exact differential \(dS\): in a reversible infinitesimal process \(dS = \delta Q_{\rm rev}/T\) with \(T\) the system’s temperature (Equation 7.6), while in any infinitesimal process \(dS\ge\delta Q/T_{\rm res}\) with \(T_{\rm res}\) the reservoir’s (Equation 7.7), reducing to \(dS\ge 0\) for an isolated system.
The work across a moving boundary is \(\delta W = p_{\rm ext}\,dV\), fixed by the boundary pressure \(p_{\rm ext}\); it equals the gas-pressure form \(p\,dV\) only when the gas is in internal equilibrium and the boundary is mechanically balanced (frictionless and force-balanced).
Combining the First and Second Laws for an infinitesimal process gives the entropy generated \(\delta S_{\rm gen} = dS - \delta Q/T_{\rm res}\ge 0\) (Equation 7.8, \(T_{\rm res}\) the reservoir’s) and the work bound \(\delta W = p_{\rm ext}\,dV \le p\,dV\) (Equation 7.9). A process is reversible iff \(\delta S_{\rm gen}=0\), i.e. \(T = T_{\rm res}\) and \(p = p_{\rm ext}\), and then the First Law collapses to the fundamental relation \(dU = T\, dS - p\, dV\) (Equation 7.10); in general \(\delta S_{\rm gen}\) splits into a thermal and a mechanical part (Equation 7.11).
Read as a relation among state functions, \(dU = T\, dS - p\, dV\) gives \(T = (\partial U/\partial S)_V\), \(p = -(\partial U/\partial V)_S\) (Equation 7.12) in the energy representation and \(1/T = (\partial S/\partial U)_V\), \(p/T = (\partial S/\partial V)_U\) (Equation 7.14) in the entropy representation, with \(1/T\) the integrating factor of \(\delta Q_{\rm rev}\). In the variables \((T,V)\) it yields the \(T\, dS\) equation \(T\, dS = n\,c_V\, dT + [(\partial U/\partial V)_T + p]\, dV\) (Equation 7.15), reproducing the ideal-gas entropy in one step.
7.8 Problems
Problem 7.1 \(2.0\ \mathrm{mol}\) of a monatomic ideal gas (\(c_V = \tfrac{3}{2}R\)) is taken from state A \((T_1 = 300\ \mathrm{K},\ V_1 = 10\ \mathrm{L})\) to state B \((T_2 = 450\ \mathrm{K},\ V_2 = 25\ \mathrm{L})\).
Using Equation 7.2, compute the entropy change \(S(B) - S(A)\).
Identify the separate contributions of the temperature change and the volume change, and state the sign of each.
Problem 7.2 Like Problem 7.1, for \(1.5\ \mathrm{mol}\) of a diatomic ideal gas (\(c_V = \tfrac{5}{2}R\)) taken from \((T_1 = 350\ \mathrm{K},\ V_1 = 20\ \mathrm{L})\) to \((T_2 = 280\ \mathrm{K},\ V_2 = 8.0\ \mathrm{L})\). Compute \(S(B) - S(A)\) and comment on the sign of the result.
Problem 7.3 \(n = 1.0\ \mathrm{mol}\) of an ideal gas expands from \(V_1\) to \(V_2 = 3V_1\) into an evacuated, rigid, thermally insulated vessel (free expansion).
Explain why the temperature is unchanged.
Compute the entropy change of the gas, \(\Delta S_{\rm gas}\), using Equation 7.1.
Compute the entropy change of the surroundings and the total entropy change \(\Delta S_{\rm univ}\), and reconcile the result with the law of increase of entropy (Equation 7.5).
Problem 7.4 Like Problem 7.3, for \(n = 0.50\ \mathrm{mol}\) expanding to \(V_2 = 2V_1\). In addition, compute the heat that would have to be supplied to carry the gas between the same two states reversibly and isothermally at \(T = 300\ \mathrm{K}\), and compare it with the heat absorbed in the free expansion.
Problem 7.5 \(n = 2.0\ \mathrm{mol}\) of an ideal gas at \(T = 350\ \mathrm{K}\), in thermal contact with a reservoir at the same temperature, is expanded from \(V_1\) to \(V_2 = 3V_1\) in three ways: (i) reversibly, with \(p_{\rm ext} = p\); (ii) suddenly against a constant boundary pressure equal to the final gas pressure, \(p_{\rm ext} = nRT/V_2\); (iii) freely, with \(p_{\rm ext} = 0\).
For each, compute the work \(W\) done by the gas and the heat \(Q\) absorbed from the reservoir.
Compute the entropy change \(\Delta S\) of the gas (the same in all three cases) and, for each, the entropy generated \(\Delta S_{\rm gen} = \Delta S - Q/T_{\rm res}\) (Equation 7.8) and the lost work \(T\,\Delta S_{\rm gen}\).
Order the three by lost work and comment.
Problem 7.6 Like Problem 7.5, for the isothermal compression of \(n = 1.0\ \mathrm{mol}\) at \(T = 300\ \mathrm{K}\) from \(V_1\) to \(V_2 = V_1/2\), comparing (i) reversible compression (\(p_{\rm ext} = p\)) with (ii) sudden compression against a constant boundary pressure equal to the final gas pressure, \(p_{\rm ext} = nRT/V_2\). For each compute \(W\), \(Q\), \(\Delta S\), and \(\Delta S_{\rm gen}\), and identify which process wastes work.
Problem 7.7 An ideal gas with constant molar heat capacity \(c_V\) is taken from state A \((T_1, V_1)\) to state B \((T_2, V_2)\).
Specialize the \(T\,dS\) equation Equation 7.15 to an ideal gas and divide by \(T\) to obtain \(dS = n\,c_V\,\dfrac{dT}{T} + nR\,\dfrac{dV}{V}\).
Integrate to obtain \(S(B) - S(A) = n\,c_V \ln(T_2/T_1) + nR\ln(V_2/V_1)\) in a single step, the result that Problem 6.13 reached by following two separate reversible paths.
Problem 7.8 (More demanding.) Like Problem 7.7, for \(n = 1.0\ \mathrm{mol}\) of a diatomic gas (\(c_V = \tfrac{5}{2}R\)) taken from \((T_1 = 300\ \mathrm{K},\ V_1 = 5.0\ \mathrm{L})\) to \((T_2 = 450\ \mathrm{K},\ V_2 = 2.0\ \mathrm{L})\).
Use the \(T\,dS\) equation to compute \(S(B) - S(A)\) in one step, and evaluate it numerically.
The reversible heat \(\delta Q_{\rm rev} = T\,dS\) is not a state function: using the two paths of Problem 6.13, show that the heat absorbed on the isothermal leg differs between them, even though \(\Delta S\) is the same.
Problem 7.9 A system has the fundamental relation, in the entropy representation, \(S(U, V) = S_0 + n\,c_V \ln(U/U_0) + nR\ln(V/V_0)\), where \(S_0\), \(U_0\), \(V_0\) are constants.
Using Equation 7.14, compute \((\partial S/\partial U)_V\) and \((\partial S/\partial V)_U\), and identify \(1/T\) and \(p/T\).
Hence show \(U = n\,c_V T\) and derive the equation of state \(pV = nRT\).
Problem 7.10 A system has the fundamental relation, in the energy representation, \(U(S, V) = U_0 \left(\dfrac{V}{V_0}\right)^{-R/c_V} \exp\!\left(\dfrac{S - S_0}{n\,c_V}\right)\).
Using Equation 7.12, compute \(T = (\partial U/\partial S)_V\) and \(p = -(\partial U/\partial V)_S\).
Show that \(T = U/(n\,c_V)\) and \(p = nRT/V\), recovering the same ideal gas as Problem 7.9, and explain why the two fundamental relations carry the same physical content. (More demanding.)
Problem 7.11 (More demanding.) Two identical bodies, each an incompressible solid of constant heat capacity \(C\) (so its entropy depends on temperature alone, \(dS = C\,dT/T\)), are initially at temperatures \(T_1\) and \(T_2\) with \(T_1 > T_2\). They are placed in thermal contact, isolated from everything else, and reach a common final temperature.
Find the final temperature \(T_f\).
Compute the total entropy change and show it is positive for \(T_1 \neq T_2\). (Hint: compare the arithmetic and geometric means of \(T_1\) and \(T_2\).)
State the entropy change of the surroundings and explain it.
Problem 7.12 A body that is an incompressible solid of constant heat capacity \(C\), initially at temperature \(T_0\), is placed in thermal contact with a reservoir at temperature \(T_{\rm res} < T_0\) and cools to \(T_{\rm res}\).
Compute the entropy change of the body.
Compute the entropy change of the reservoir, which absorbs the heat \(Q = C(T_0 - T_{\rm res})\) at the fixed temperature \(T_{\rm res}\).
Show that the total entropy change is positive. (More demanding.)
Problem 7.13 Consider an infinitesimal process of a closed system that is in internal equilibrium (so its temperature \(T\) and pressure \(p\) are defined), in contact with a reservoir at temperature \(T_{\rm res}\) and delivering boundary work against \(p_{\rm ext}\).
Starting from \(T\,dS = dU + p\,dV\) and the First Law \(dU = \delta Q - p_{\rm ext}\,dV\), show that \(\delta Q = T\,dS - (p - p_{\rm ext})\,dV\) and \(\delta W = p\,dV - (p - p_{\rm ext})\,dV\), and that the common shortfall cancels in \(dU = \delta Q - \delta W\).
Hence derive the split \(\delta S_{\rm gen} = \delta Q\,(1/T - 1/T_{\rm res}) + (p - p_{\rm ext})\,dV/T\) (Equation 7.11), and explain why each term is non-negative. (More demanding.)
Problem 7.14 (More demanding.)
Using the force balance on a piston sliding with kinetic friction (Figure 7.5), show that on both expansion and compression \((p - p_{\rm ext})\,dV \ge 0\), and hence that the boundary work satisfies \(\delta W = p_{\rm ext}\,dV \le p\,dV\) (Equation 7.9).
From the Clausius inequality in differential form (Equation 7.7), show that for a thermally isolated system \(dS \ge 0\), with equality if and only if the process is reversible.
Problem 7.15 (Forward-looking: previews the van der Waals gas, developed in the guided problem of Lecture 9.) A gas obeys the co-volume equation of state \(p = \dfrac{nRT}{V - nb}\) (\(b\) a constant) and has \((\partial U/\partial V)_T = 0\).
Using the \(T\,dS\) equation Equation 7.15, write \(T\,dS\) in terms of \(dT\) and \(dV\).
Taking \(c_V\) constant, integrate to find \(S(T_2, V_2) - S(T_1, V_1)\).
For the same temperatures and volumes, compare the volume contribution with that of an ideal gas, and state which is larger for an expansion.
Problem 7.16 A van der Waals gas has \((\partial U/\partial V)_T = a n^2/V^2\) (\(a\) a constant) and the equation of state \(p = \dfrac{nRT}{V - nb} - \dfrac{a n^2}{V^2}\).
Using Equation 7.15, show that the \(a\) terms cancel and \(T\,dS = n\,c_V\,dT + \dfrac{nRT}{V - nb}\,dV\).
Taking \(c_V\) constant, integrate to find \(\Delta S\), and note that it depends on \(b\) but not on \(a\). (More demanding. Forward-looking: previews the van der Waals gas, developed in the guided problem of Lecture 9.)
Problem 7.17 (More demanding.) A rigid, thermally insulated cylinder of cross-section \(A\) contains \(n\) moles of an ideal gas (constant molar \(c_V\)), closed by a piston that is an incompressible solid of constant heat capacity \(C\). The piston slides against the cylinder wall with sliding (kinetic) friction of magnitude \(f_k\), and the static friction can take any value up to a maximum \(f_s \ge f_k\). Take the thermodynamic system to be the gas together with the piston, which we call the composite system. The cylinder is rigid, adiabatic, and of negligible heat capacity, so all the frictional heat is retained in the gas and the piston, which stay in thermal contact at a common temperature \(T\). The composite system exchanges no heat with the outside, and the only energy crossing its boundary is the work the external agent does on the piston.
Explain why the piston’s entropy depends on its temperature alone, \(S_{\rm pis}(T) = C\ln(T/T_0)\), while the gas has \(S_{\rm gas}(T, V) = n\,c_V \ln(T/T_0) + nR\ln(V/V_0)\) (both relative to a reference state \((T_0, V_0)\)). Write the entropy of the composite system, \(S = S_{\rm gas} + S_{\rm pis}\).
State the conditions for the gas and the piston to be in mutual equilibrium. Show that, because the static friction can take any value up to \(f_s\), mechanical equilibrium of the piston holds for a band of gas pressures \(|p - p_{\rm ext}| \le f_s/A\), so the volume is not fixed by mechanical equilibrium alone; only for \(f_s = 0\) does \(p = p_{\rm ext}\).
Take \(f_s = f_k = 0\) first. The agent compresses the gas reversibly and adiabatically from the initial state \((T_1, V_1)\) to volume \(V_2 < V_1\). Show that \((n\,c_V + C)\,dT/T = -nR\,dV/V\) along the process. Introducing the adiabatic index of the composite system, \(\gamma' = 1 + \dfrac{nR}{n\,c_V + C}\), this integrates to the adiabat \(T\,V^{\gamma'-1} = \mathrm{const}\), so that \(T_f^{\rm rev} = T_1 (V_1/V_2)^{\gamma'-1}\); verify that the total entropy change is zero.
Now restore the friction and compress the gas to the same volume \(V_2\). Show, with no integration, that the work dissipated by friction is \(W_{\rm diss} = (f_k/A)(V_1 - V_2)\).
Without yet finding \(T_f\), show that the total entropy change is \(\Delta S = (n\,c_V + C)\ln(T_f/T_f^{\rm rev})\). (The actual and the reversible process start from the same state and end at the same volume \(V_2\), differing only in the final temperature; since \(S\) is a state function and the reversible process of (c) has \(\Delta S = 0\), the volume term cancels.) Conclude from the law of increase of entropy (Equation 7.5) that \(T_f \ge T_f^{\rm rev}\), with equality only when \(f_k = 0\).
To find \(T_f\) itself, apply the First Law to the composite system along the stroke and show that \[(n\,c_V + C)\,dT = -\left(\frac{nRT}{V} + \frac{f_k}{A}\right)\,dV .\] Solve this linear equation for \(T_f\), and confirm that it exceeds \(T_f^{\rm rev}\) by an amount governed by \(f_k\). (Hint: write it as \(\dfrac{dT}{dV} + (\gamma'-1)\dfrac{T}{V} = -\dfrac{f_k}{A(n\,c_V + C)}\) and multiply by the integrating factor \(V^{\gamma'-1}\), so the left-hand side becomes \(\dfrac{d}{dV}\big(T\,V^{\gamma'-1}\big)\); then integrate from \(V_1\) to \(V_2\).)
Problem 7.18 (More demanding.) Like Problem 7.17, but the agent lets the gas expand adiabatically from \(V_1\) to \(V_2 > V_1\) against friction. Use \(n = 1.0\ \mathrm{mol}\) of a monatomic gas (\(c_V = \tfrac{3}{2}R\)), a piston of heat capacity \(C = 2R\) and area \(A = 0.0100\ \mathrm{m^2}\), kinetic friction \(f_k = 600\ \mathrm{N}\), and initial state \(T_1 = 400\ \mathrm{K}\), \(V_1 = 10.0\ \mathrm{L}\), \(V_2 = 20.0\ \mathrm{L}\).
Compute the dissipated work \(W_{\rm diss} = (f_k/A)(V_2 - V_1)\), and the frictionless final temperature \(T_f^{\rm rev}\) (part (c) of Problem 7.17); confirm the gas would cool.
Show that the First Law for the composite system gives \((n\,c_V + C)\,dT = -\left(\dfrac{nRT}{V} - \dfrac{f_k}{A}\right)\,dV\), solve for \(T_f\) (integrating factor \(V^{\gamma'-1}\), as in Problem 7.17), and compute the total entropy change \(\Delta S = (n\,c_V + C)\ln(T_f/T_f^{\rm rev})\) (the volume term cancels as in Problem 7.17(e)), verifying \(\Delta S > 0\).
Problem 7.19 A gas is enclosed in a cylinder by a piston of area \(A\). For each situation below, identify the system boundary, state the boundary pressure \(p_{\rm ext}\) and whether it equals the gas pressure \(p\), and give the work \(\delta W\) the system delivers (Figure 7.3, Figure 7.4, Figure 7.5).
System = the gas alone; the piston is withdrawn slowly, quasi-statically.
System = the gas alone; the piston is withdrawn faster than the gas can follow.
System = gas \(+\) piston, with an ideal frictionless piston moved quasi-statically.
System = gas \(+\) piston, the piston sliding with kinetic friction of magnitude \(f\), in quasi-static expansion.
The same as (d), in quasi-static compression. Explain why this process is irreversible even though it is quasi-static.
Problem 7.20 (More demanding.) Like Problem 7.19(d,e): a piston of area \(A = 0.0100\ \mathrm{m^2}\) slides with kinetic friction of magnitude \(f = 40\ \mathrm{N}\), and the gas, in internal equilibrium at pressure \(p\), drives it quasi-statically (Figure 7.5).
On expansion, express the boundary pressure \(p_{\rm ext}\) in terms of \(p\), \(f\), and \(A\); repeat for compression.
For \(p = 1.50\times 10^{5}\ \mathrm{Pa}\), compute \(p_{\rm ext}\) in each case.
The piston is moved out by a volume \(\Delta V = 0.50\ \mathrm{L}\) and then back to its starting position, with \(p\) essentially constant. Compute the work the gas does on the piston, the work delivered to the external agent, and the energy dissipated by friction over the round trip, and verify that the dissipated energy equals \(2f\,\Delta x\), where \(\Delta x = \Delta V/A\) is the piston displacement.